Computing LD in R
Suppose you have a matrix of 0’s and 1’s representing haplotypes at a list of SNPs (or other biallelic variants) and you want to compute pairwise LD statistics in R. Computing 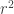
r = cor( t( data ) )
and then square it (I am assuming the SNPs are in rows and chromosomes in columns, the way I usually arrange things. If not the transpose isn’t needed.) However, computing 
compute.ld <- function( haps, focus.i = 1:nrow(haps), variant.names = rownames( haps ) ) {
# haps is a 0-1 matrix with L SNPs (in rows) and N haplotypes (in columns).
# Since the values are 0, 1, we rely on the fact that a*b=1 iff a and b are 1.
# assume focus.i contains l values in range 1..L
L = nrow( haps )
l = length( focus.i )
# p11 = lxL matrix. i,jth entry is probability of 11 haplotype for ith focal SNP against jth SNP.
focus.hap = haps[ focus.i, , drop = FALSE ]
p11 <- ( focus.hap %*% t( haps )) / ncol( haps )
# p1. = lxL matrix. ith row is filled with the frequency of ith focal SNP.
p1. <- matrix( rep( rowSums( focus.hap ) / ncol( haps ), L ), length( focus.i ), L, byrow = FALSE )
# p.1 = lxL matrix. jth column is filled with the frequency of jth SNP.
frequency = rowSums( haps ) / ncol( haps )
p.1 <- matrix( rep( frequency, length( focus.i ) ), length( focus.i ), L, byrow = TRUE )
# Compute D
D <- p11 - p1. * p.1
# Compute D'
denominator = pmin( p1.*(1-p.1), (1-p1.)*p.1 )
wNeg = (D < 0)
denominator[ wNeg ] = pmin( p1.*p.1, (1-p1.)*(1-p.1) )[wNeg]
denominator[ denominator == 0 ] = NA
Dprime = D / denominator
# Compute correlation, this result should agree with cor( t(haps ))
R = D / sqrt( p1. * ( 1 - p1. ) * p.1 * ( 1 - p.1 ))
R[ frequency[ focus.i ] == 0 | frequency[ focus.i ] == 1, frequency == 0 | frequency == 1 ] = NA
if( !is.null( variant.names )) {
rownames( D ) = rownames( Dprime ) = rownames( R ) = variant.names[ focus.i ]
colnames( D ) = colnames( Dprime ) = colnames( R ) = variant.names
names( frequency ) = variant.names
}
return( list( D = D, Dprime = Dprime, frequency = frequency, R = R ) ) ;
}
Big deal.
Well, what’s interesting about this function is that it’s quite fast (relatively speaking):
> dim(A)
[1] 1002 1000
> system.time( { r = cor( t(A)) } )
user system elapsed
1.268 0.020 1.296
> system.time( { d = compute.ld( A ) } )
user system elapsed
1.340 0.128 1.154
…which says that, on this dataset of 1002 SNPs and 1000 samples, compute.ld() takes less time than cor(), despite computing more stuff.
You can also use it to compute LD between a subset of SNPs and all the others:
> d = compute.ld( A, focus.i = c( 500,501 )) > dim(d$Dprime) [1] 2 1002
Example
Aha, you say, but does it work?. One way to test it is to try about the simplest set of haplotypes that give interesting results – so I tested it on every possible combination of five chromosomes. (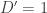
data = as.matrix( expand.grid( c(0,1),c(0,1),c(0,1),c(0,1),c(0,1) ) ) ld = compute.ld( data ) Dprime = ld$Dprime R = ld$R
The function computes D, correlation (R), and D’. Let’s check it gets R right first:
max( cor( t( data ) ) - R ), na.rm = T ) [1] 8.881784e-16
So this agrees with cor() although there is some numerical error. Also, there’s a symmetry in the data, namely ![\text{data}[i,] = 1 - \text{data}[32-i,]](https://s0.wp.com/latex.php?latex=%5Ctext%7Bdata%7D%5Bi%2C%5D+%3D+1+-+%5Ctext%7Bdata%7D%5B32-i%2C%5D&bg=ffffff&fg=333333&s=0&c=20201002)
reversed = 32:1 stopifnot( is.na( Dprime) == is.na( Dprime[reversed, reversed])) stopifnot( is.na( Dprime ) == is.na( Dprime[reversed,])) stopifnot( is.na( Dprime ) == is.na( Dprime[, reversed])) max( ( Dprime - Dprime[reversed,reversed] ), na.rm = T ) [1] 4.218847e-15 max( ( Dprime + Dprime[reversed,] ), na.rm = T ) [1] 3.330669e-16 max( ( Dprime + Dprime[,reversed] ), na.rm = T ) [1] 3.330669e-16
Also,
wNA = which( rowSums( data ) == 0 | rowSums( data ) == ncol( data ) ) stopifnot( length( which( !is.na( Dprime[wNA,wNA] ) ) ) == 0 ) stopifnot( length( which( is.na( Dprime[-wNA,-wNA] ) ) ) == 0 )
To test that the values are actually correct, let’s figure out what the answer should be. With 5 chromosomes, possible nonzero frequencies are 




w = which( rowSums( data ) == 1 | rowSums( data ) == 4 ) table( compute.ld( data, focus.i = w )$Dprime )
which gives
-1 1 150 150
Suppose then that both SNPs have frequency 
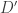
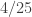





rownames( data ) = paste( data[,1], data[,2], data[,3], data[,4], data[,5], sep = '' ) w = which( rowSums( data ) == 2 ) compute.ld( data[w,] )$Dprime
which outputs
11000 10100 01100 10010 01010 00110 10001 01001 00101 00011 11000 1.0000 0.1667 0.1667 0.1667 0.1667 -1.0000 0.1667 0.1667 -1.0000 -1.0000 10100 0.1667 1.0000 0.1667 0.1667 -1.0000 0.1667 0.1667 -1.0000 0.1667 -1.0000 01100 0.1667 0.1667 1.0000 -1.0000 0.1667 0.1667 -1.0000 0.1667 0.1667 -1.0000 10010 0.1667 0.1667 -1.0000 1.0000 0.1667 0.1667 0.1667 -1.0000 -1.0000 0.1667 01010 0.1667 -1.0000 0.1667 0.1667 1.0000 0.1667 -1.0000 0.1667 -1.0000 0.1667 00110 -1.0000 0.1667 0.1667 0.1667 0.1667 1.0000 -1.0000 -1.0000 0.1667 0.1667 10001 0.1667 0.1667 -1.0000 0.1667 -1.0000 -1.0000 1.0000 0.1667 0.1667 0.1667 01001 0.1667 -1.0000 0.1667 -1.0000 0.1667 -1.0000 0.1667 1.0000 0.1667 0.1667 00101 -1.0000 0.1667 0.1667 -1.0000 -1.0000 0.1667 0.1667 0.1667 1.0000 0.1667 00011 -1.0000 -1.0000 -1.0000 0.1667 0.1667 0.1667 0.1667 0.1667 0.1667 1.0000
…so I basically think it does work.
